User Guide
Overview
CrustyBase is a repository and analysis suite for crustacean
transcriptome data. Each dataset describes the activity of
every expressed gene in a particular species, across a set of
samples.
CrustyBase currently contains 37 transcriptome
datasets, but anyone can add to this in the future by uploading
their own.
You can access the datasets on CrustyBase through different tools (aka apps). We may implement more apps depending on user feedback but for now, there are two core apps:
While the number of transcriptomes is small, navigating datasets remains quite
simple. But once the database grows, finding a relevant dataset among thousands
becomes challenging. The browser tool was designed to efficiently find
the datasets that are of most interest to you.
You can search for
a particular species, or try any other keyword that is relevant to you.
"Molt", "disease", "virus", "immune" and "brain" should all return related
datasets. You can also search by taxonomy ("portunidae").
Each dataset has a dedicated page describing the species, data
and experimental conditions.
The BLAST tool is a long-time staple of the bioinformatics toolbelt. Submit
a DNA or protein sequence that you're interested in and it will give you back the
best-matching sequences in the transcriptomes that you have selected.
Typically you would
obtain the sequence of a gene of interest from a related species (there
are millions available on NCBI) and use that as a query sequence.
Learn more about the NCBI's BLAST tool
here.
Once your result has been returned, you can expand details on the
datasets that yield the most interesting results. You
can then click (or use arrow keys) through the matching transcripts
while viewing expression profiles and sequence structures in real time.
Datasets
Datasets have been structured to provide as much information as possible for each transcriptome.
Metadata
Each dataset is described by a structured array of metadata,
including information like taxonomy, experiment description and
assembly procedure. It can also include a reference to a publication
so that the dataset author can be easily cited, which we
strongly encourage if you find a dataset useful.
You can view the metadata for a dataset on its profile page in
the data browser.
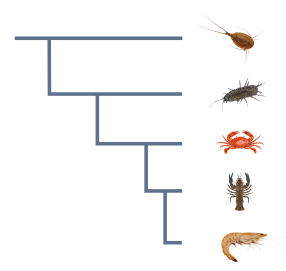
Transcript data
Each dataset has a number of data types corresponding to each transcript. Some of these are rendered in a graphical display as you browse through transcripts in a BLAST result, and all of them are available to download in some form.
These data types are summarised in the table below.
| Data type | Description | Rendered in output | Available for download |
|---|---|---|---|
| Nucleotide sequence | cDNA sequence of the transcript. This sequence is the origin of all other data types. | Only as alignment | Yes, full access required |
| CDS sequence | Coding DNA sequence predicted by TransDecoder. | No | Yes, full access required |
| Peptide sequence | Protein sequence predicted by TransDecoder. | Only as alignment | Yes, full access required |
| Expression data | Mean expression level across experiment features | Yes | Yes, full access required for raw data |
| Conserved domains | Conserved protein domains predicted by CD-search | Yes | Yes, graphics only |
BLAST search
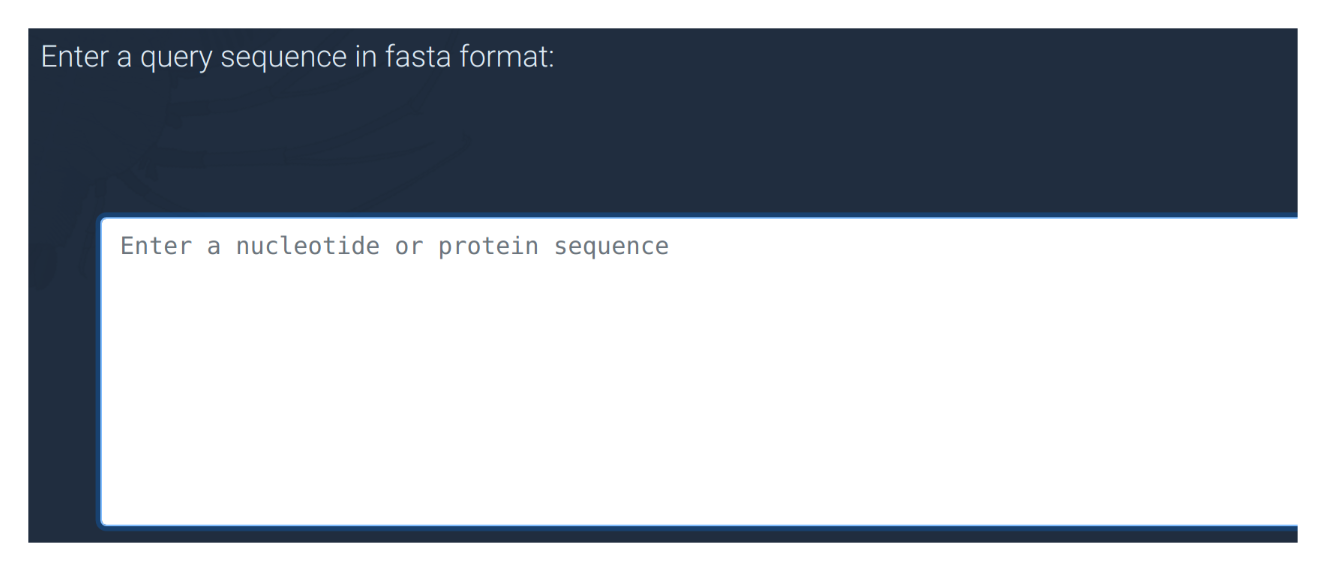
Search for a sequence
The BLAST submission form is made up of three simple components:
Query sequence input
To conduct a BLAST search you must first find a DNA or protein sequence of a gene that you're interested in. The "sample sequence" button can give you an example if you're just trying it out. Sequences can be obtained from the NCBI website or sometimes directly from published research articles. Copy and paste your sequence into this box to begin. It is worth remembering that protein sequences are usually more conserved than DNA between distant taxa, and are therefore more likely to match your target gene.
Database list
Select the transcriptome dataset you wish to search from this list. There
may be many databases, but only 10 are shown at a time. To find the
database you're looking for, simply type in some keywords into the
input field above to filter the database list. Try searching with
species or family names, tissues ("brain" or "gill") or other
biological keywords such as "immune", "environment" or "reproduction".
If you want to search and view the datasets in more detail, switch
to the data browser.
The access level of each database is shown on the right as a
green (full access) or orange (restricted access) light.
You can add one or many datasets to the "selected" pane before
running the search, but more datasets will of course increase
the run time.
Search algorithm
The search algorithm that you choose depends on the query sequence
that you entered above:
- BLASTN searches nucleotide transcriptomes with a DNA sequence
- tBLASTN takes a protein sequences and searches translated nucleotide sequences on-the-fly. This tends to be more reliable when searching cross-genus and beyond, as protein sequences are almost always more conserved.
Viewing the result
- Datasets with transcripts matching your query are stacked up the page.
- Each dataset in the stack features a species image, experiment description and table summarising the BLAST hits.
- Select transcripts in the table with the checkbox, and download data relating to them.
- Click "expand" to open up a full-screen view of a dataset's results
- Click or use arrow keys to visualize transcripts in three corresponsing panes:
The alignment pane
A conventional alignment out from the BLAST tool is displayed in
the first viewing pane. This shows exactly which residues match
between the query sequence and the selected transcript. Match
statistics are shown above the alignment. One match may return
a number of HSPs (high-scoring pairs).
HSPs are sections of matching sequence. When searching a
transcriptome with protein or cDNA sequence you would normally
expect to find one HSP. Two or more indicates large insertions
or deletions between the query and subject sequence, probably
due to differential mRNA splicing or perhaps a sequencing error.

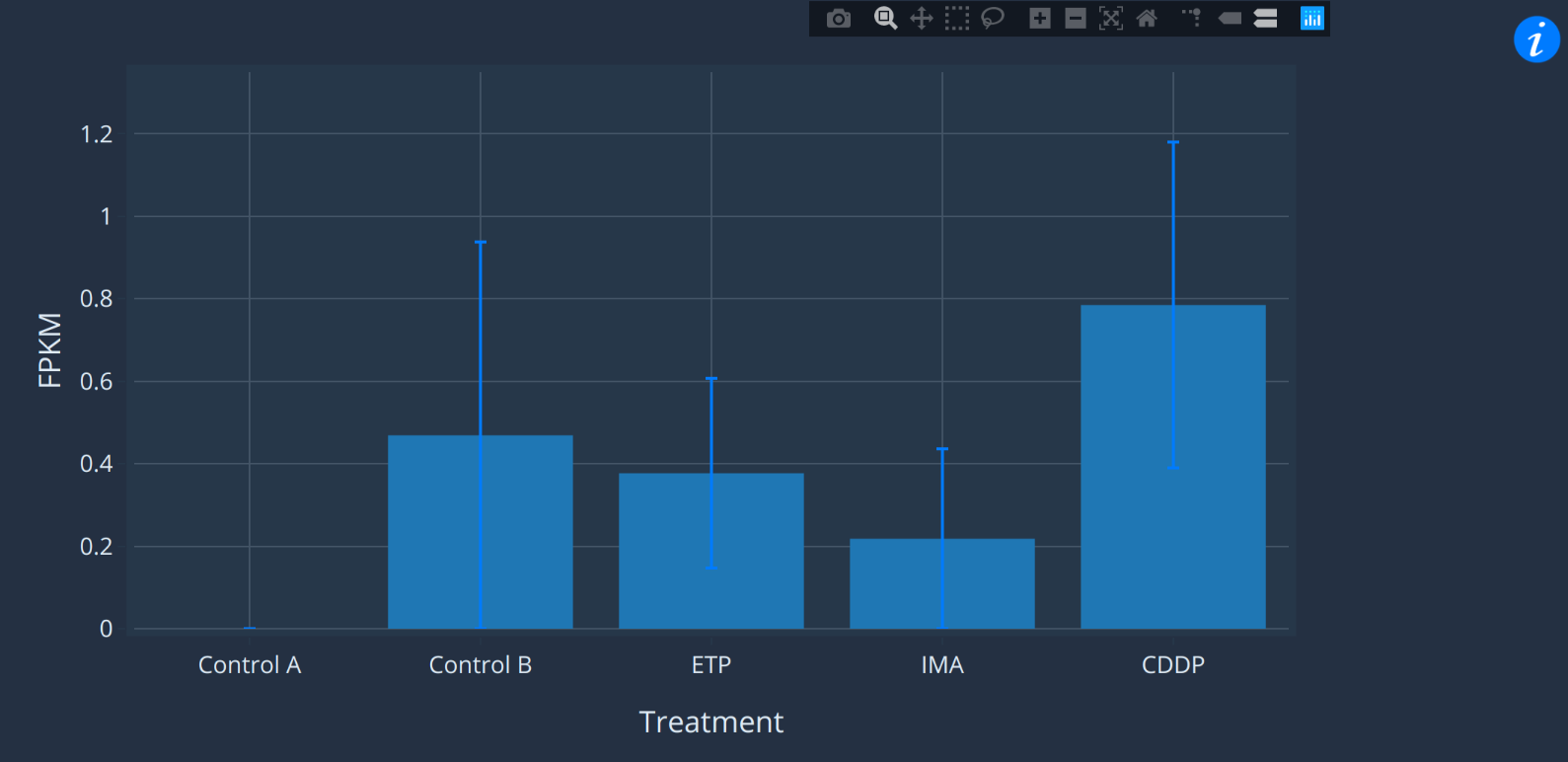
The expression pane
The expression pane shows the expression profile of the
selected transcript. Move the cursor over markers on the chart
to see mean and standard error of the data. If you aren't sure
what the x-axis labels mean, try hovering over the info symbol
on the right to see the experiment description. You can also
click-and-drag on the y-axis to increase or decrease scale.
This can be useful for zooming in on low-expressed samples.
You may notice that some datasets are displayed with bar
charts and others with line graphs. This depends on whether
the expression data describes a categorical variable
(tissues, treatment etc.) or continuous variable (days,
temperature etc.).
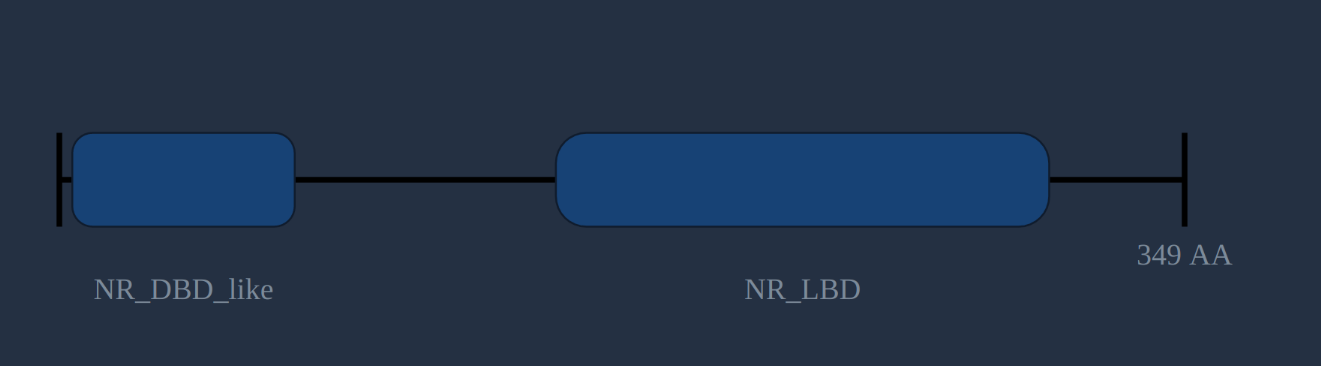
The protein structure pane
The structure pane shows the protein length predicted by TransDecoder, with conserved domains (predicted by the NCBI's CD-search ) plotted along its length. Domains are linked to their descriptions in NCBI, PFAM, TIGRFAM and other databases which can be followed by clicking on them. Not all transcripts will have a predicted protein, and not all predicted proteins will have predicted domains. However, if you know what structure to expect (like the nuclear receptor DBD and LBD in the protein on the right) it can be a good indication of a correct and complete sequence.
Downloading sequence data
Plots and sequences from the results page can be downloaded in bulk.
Select transcripts of intereset in the match list
As you browse matching transcripts, you can select any that
seem interesting with the checkboxes on the right of the match
list.
Once you're happy you've got all the interesting matches, click
the download button above to open the download dialog.
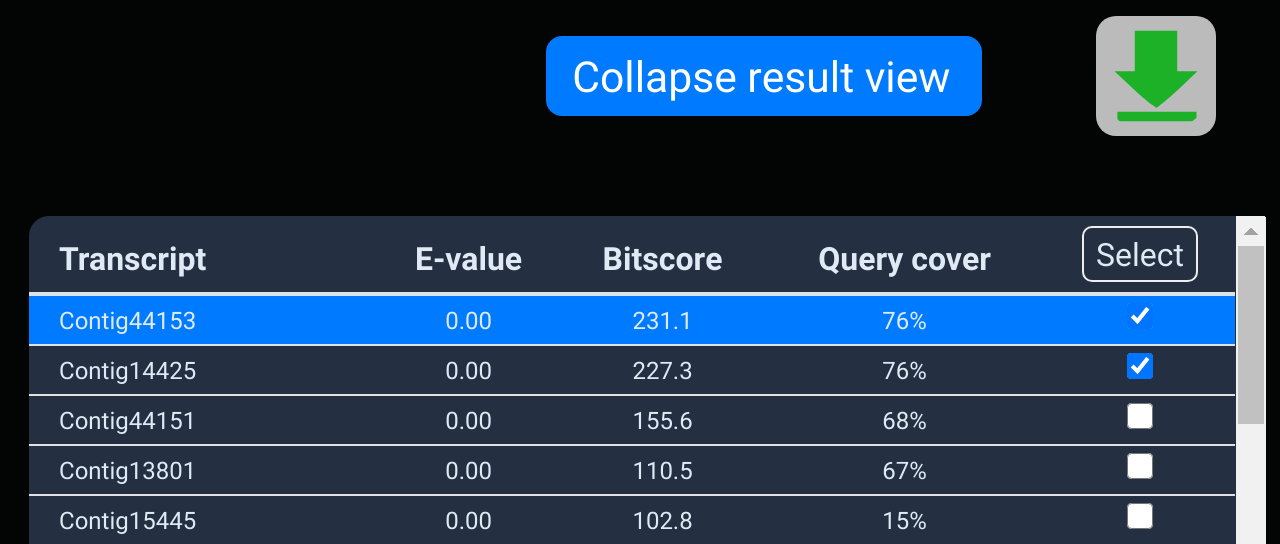
The download dialog
Simply select the formats that you wish to download and click
the download button. The available formats depend on whether
the owner of the dataset allows full or only
partial access.
It may take up to several minutes to render the requested
files if many formats and transcripts are selected.
You optionally add a file prefix. This will be used to name the
downloaded file, so you can remember the origin of these files
later on. For example, entering "myresult" will lead to
downloading a file called "myresult.zip".
Saving results
If you are a registered user, you have the option to save results
that might be useful in the future. When logged in,
you should see a "save" button in the top-right. Click save,
enter a useful identifier (so you can remember what it is) and
then click save or hit the enter key.
You can then return to this result at any time through the
saved results
page of your user profile. CrustyBase will store a maximum of
200 saved results. When this limit is reached, further
saves will begin overwriting the oldest save.
You can also view and revisit all results from the past 7 days
in the "BLAST history" panel at the bottom of your
dashboard.
User accounts
You don't need to be registered to use CrustyBase, but it does come with benefits!
When you create an account with CrustyBase, you will be able to save BLAST results and view your search history. You can also create groups and upload your own transcriptome datasets. It's free to register and always will be.
Account overview
The
dashboard
gives you a brief overview of your account. This page allows
you to edit your personal details,
view your groups and datasets, view recent search history and
delete your account (why would you do that!?).
When you are logged in, the dashboard and other
pages related to your account can be found by clicking on the
login prompt in the top-right of any every page, as shown on the
right.
Groups
Groups control access to transcriptome datasets.
There are two situations when you might need to make use of groups:
- You are going to import a dataset
- You want to get access to a colleague's datasets
You can manage groups through your user profile.
Data access
The purpose of groups is to control data access.
Not all datasets are fully accessible to the public, but the members
of a group always have full access to that group's data. Groups
are designed to reflect data ownership in the real world - datasets
are typically owned by a research group, not by a single person in
that group.
You can still view and search restricted datasets,
but you cannot access or download
raw sequence or expression data. The purpose of this is to
encourage sharing of datasets which are restricted by intellectual
property rights, since graphical results are usually
insufficient for published research.
If you find something of interest in these
datasets, we encourage users to look up the owner of the dataset
and seek collaboration. You can find out who uploaded the
dataset by checking the dataset's profile in the
data browser.
Create a group
The only time you might need to create a group is when you are
going to import a dataset. You can also consider joining a
colleague's group and importing the data to there, if you wish to
share access. However, if you are going to import publicly available
data, you can simply opt to import the dataset into the
Public Domain instead.
You can create new groups in the
group management
page, which is accessible through the login prompt in the top-right.
When creating a group, think of a name that is descriptive and
unique. For example, Albert Einstein's research group at ETH
Zurich might be called "Einstein ETHZ".
Join a group
If a colleague has a research group that you wish to join, they
will need to send you an invite. This can be done easily by
visiting the
group management page.
Simply select the appropriate group and hit the "invite" button.
Enter the email address of the person you wish to invite and
they will be sent a link to join the group.
Check the email address carefully - once someone joins there is
no (easy) way to remove them from the group.
Leave a group
There are situations where you may want leave a group - perhaps
the group is redundant or you are leaving an institution. Simply
select the group on the
group management
page and select "leave group" at the bottom of the page.
Consider this carefully though. Once you leave you'll lose access
to the group's datasets and, if you were the last member in the
group, the group and all its data will be deleted. This is the
only way to delete a group.
Import data
Why import data?
There are a number of reasons to import data into CrustyBase.
The most obvious reason to contribute your data is because it is
the right thing to do! We are all in the business of advancing
global knowledge, and the process is far more efficient when we
work together.
Aside from that, there several benefits of having your
data in the hands of CrustyBase:
- CrustyBase is a free service for navigating your transcriptome data.
- You and your research group can access your data wherever you are.
- Increase your exposure. Make collaborations, get citations.
Prepare an import
You can find the
data import app
in the data tab in the navigation bar.
There are three phases to the data import process:
- Metadata input (dataset information and access level)
- File upload & validation
- Review & submit
Public access level
Full access allows any user of CrustyBase to download raw sequence and expression data from any transcripts that they find. This data might be sufficient to publish findings on CrustyBase.
Partial access restricts the data types that a public user is able to download, allowing only graphical content to be seen. Given that publication typically requires reporting of original data, users who wish to make such use out of these datasets would be expected to contact the dataset owner to seek collaboration.
All members of the group owning the dataset have full access, including permission to edit and delete the dataset.

File upload
Upload files must be
correctly formatted for the server to parse them correctly. If
there is a problem validating the data, you will be given a
useful message to help fix the problem. It should be possible
to make any formatting changes with conventional spreadsheet
and text-editor software.
There are two files that need to be uploaded:
- Transcriptome assembly in FASTA format
- Expression data in CSV format
These files are limited to 1000MB each.
Assembly file
The assembly file should be a FASTA-formatted sequence file.
Each sequence should start with a title line, beginning with
an angle bracket ">" and a new line. The following sequence
should be composed of only the characters "ATGCN" and new lines.
The title can be no longer than 25 characters for any given
sequence - any longer than this is unnecessary and makes them
difficult to display on CrustyBase.
In the FASTA
file on the right you can see that the first sequence title (green)
is an appropriate length and format, whereas the second sequence
title (red) has been appended with surplus information by the
assembly software. This additional information needs to be
removed.
If you are unable to reformat the sequence titles,
try uploading the file with the "Reformat contig IDs" option
enabled. Please note that you will have no way to link CB
results to your local dataset if you choose this method, as
the contig IDs will no longer match.
CGACACCCAGAAGGGCCTGCAGCACGCCATGATGCAGATGAACGGCCCGATGATGGAAGG ACGTCGCCTGGATCTGCGCGATGATCCCGCATCACATGGGCGCCATCGCCATGGCCCAGG AATCCCGAGGCTAAGAAGATCGCCGAGAAGAGCATCCAGGAACAGGAGAAGAGCATCAAG
>comp3_c0_seq1 len=319 path=[12086:0-74 3106:75-318]
ATCTGTTTCTCCTTTTCATATTTTTCTTTTCTTTTGTTCCCTGTGTTCCACTTCTCTGTC CTTTCACTTCCCTCCTTCTCTTCCTTCTGTTTATTTGCTTCTTCTTCAGTATCCTTTCTT CCTCTTTTCCTTTTCCCTTCGCATCTTTCTCTCCTTCTTCTTTCTCTTCATCTATTCCTT
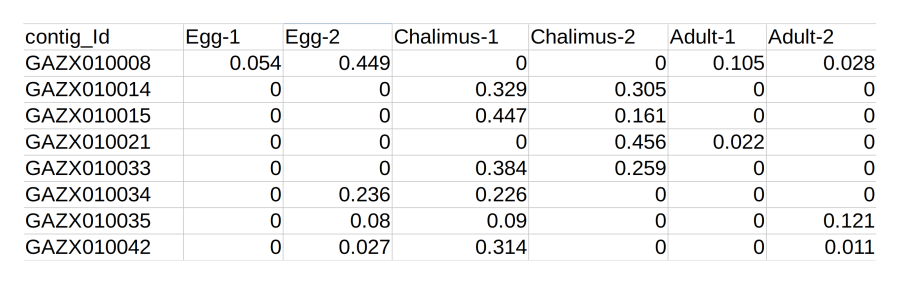
Expression file
The expression quantitation file should be a plain CSV file.
If the samples were sequenced with replicates, makes sure that
this file includes the raw replicate data and not mean data.
The file on the right shows how the data should be formatted.
Contig IDs in the first column should match those in the
assembly file, otherwise there is no way to connect the two
together!
It may be helpful for you to label the column header with
meaningful names, but these will be discarded when the dataset
is imported. Column names are taken from the "Series labels"
field during Metadata entry.
We highly recommend that you use RLE, TMM or TPM as
units of transcript expression, though there are other
metrics that can be used. For more accurate quantitation it is
often better to map reads to the CDS (coding DNA sequence)
rather than the entire transcript, as this results in a more
uniform distribution of read mapping.
Restrictions
There are several restrictions on data imports which may prevent some datasets from being imported.
Comparable samples
The transcriptome must have originated from sequencing multiple
samples that can be compared. Some transcriptomes are generated
with only one sample feature without examining any variables -
these datasets are not suitable for CrustyBase because there is
no expression data to compare. An example would be a
transcriptome of brain tissue which includes no other
features for comparison.
CrustyBase is designed to show experiments where transcript
abundance has been estimated across a number of features, for
example brain, gonad and muscle tissues. In this case, we would
be able to visualise the difference in transcript abundance
between these three tissues.
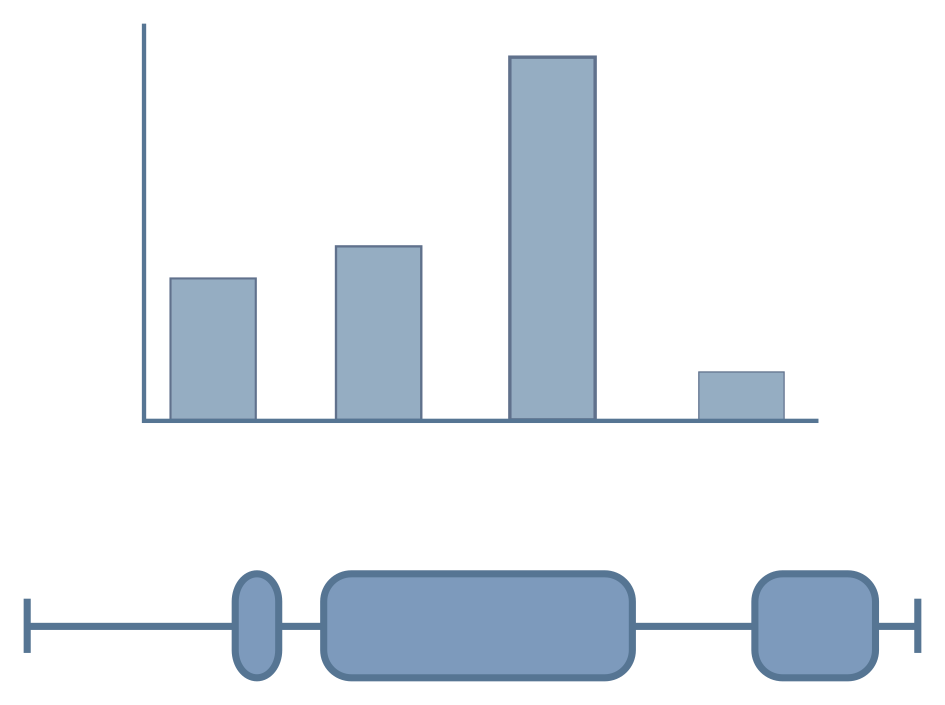
Feature levels
Features describe the variable(s) compared in an RNA-seq experiment.
Example features could be tissue type, developmental stage
or experimental treatment. However, some experiments define
multiple feature levels.
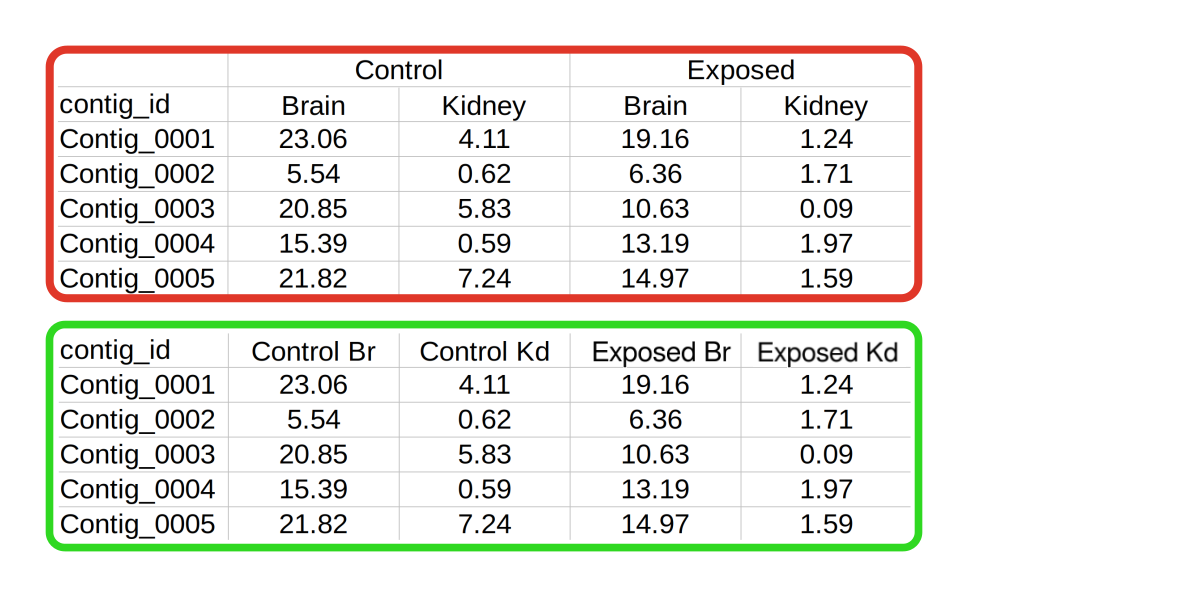
An example of this is shown on the right (red box), where samples have been taken from two different tissues under a control and treatment condition. In order to import this dataset, these features would need to be flattened into one level, as shown on the bottom panel (green box).
File size
Uploads have been restricted to a maximum file size of 1000MB to preserve server resources. If this limit prevents you from uploading a genuine dataset, please let us know by leaving some feedback . We can raise this limit if necessary.

Delete a dataset
There are a number of reasons why you might want to remove a dataset from CrustyBase. Any member of a group can delete any dataset under that group's ownership. Find the dataset under My datasets. On the profile page you will find the option to delete at the end of the Details section.
